
ROSE 软件挖掘「超级增强子」实战与代码分享
一、ROSE 软件是什么?
ROSE(Rank Ordering of Super-Enhancers)由 MIT Richard A. Young 实验室开发,是目前引用率最高的超级增强子预测工具。
rose软件下载地址:https://bitbucket.org/young_computation/rose/
软件分析原理:
-
先用 H3K27ac(或 MED1/BRD4 等)ChIP-seq peaks 定义“增强子”区间;
-
将彼此 ≤12.5 kb 的增强子“缝合”(stitched)成连续区域;
-
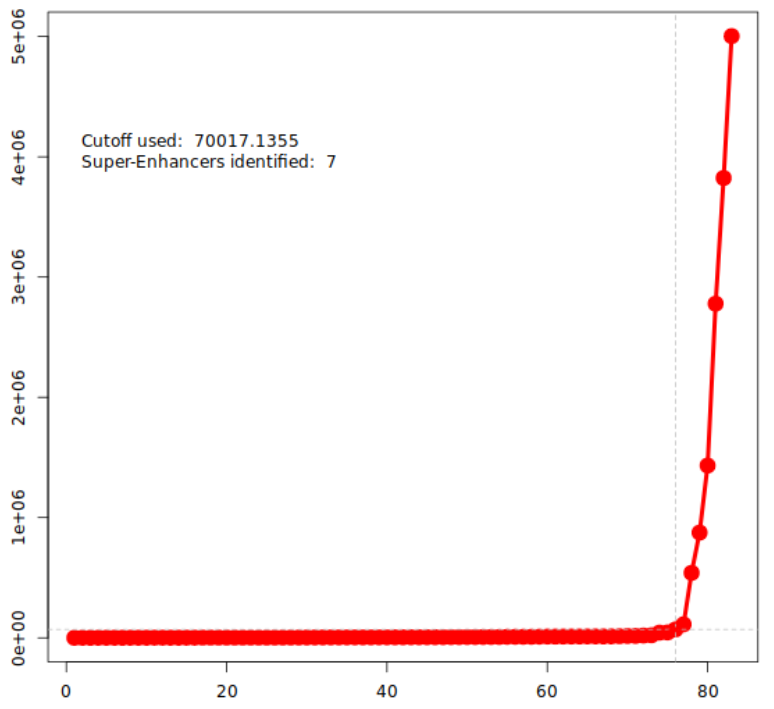
根据信号强度排序,拐点(inflection point)以上即为超级增强子,以下为普通增强子(typical enhancer)。
二、安装步骤与环境依赖
- 安装系统:Linux(CentOS/Ubuntu)
- 环境依赖:
- Python 2.7(注:不可使用 Python3环境,会报错!)
- R ≥ 3.6(用于绘图)
- SAMtools 0.1.18(新旧版亦可,需支持
sort -o)
一键下载:
cd ~ #回到Home
wget https://bitbucket.org/young_computation/rose/get/feb35cb1d955.zip #下载
unzip feb35cb1d955.zip #解压
mv young_computation-rose-* ROSE #重命名文件夹
export PATH=$PWD/ROSE/bin:$PATH ##写入环境三、输入文件准备
| 文件 | 说明 | 工具/注意事项 |
|---|---|---|
| bam | ChIP 样本(H3K27ac) | bam文件需要有.bai索引文件 |
| control bam(可选) | Input 或 IgG 对照 | bam文件需要有.bai索引文件 |
| gff/bed | 增强子初筛结果 | 通常用 MACS2 鉴定的 broad peaks |
注意:软件默认支持人(hg19 / hg38)和鼠(mm9 / mm10)的参考基因组。
四、一条命令跑完 SE 鉴定
python ROSE_main.py \
-g hg38 \
-i sample_peaks.gff \
-r sample.bam \ #需鉴定的样本
-c input.bam \ #control bam
-o ROSE_out \ #输出文件名
-s 12500 \
-t 2500- 常用参数:
-g:基因组版本(内置 hg19/hg38/mm9/mm10)-s:缝合距离,默认 12.5 kb-t:排除 TSS ±2–3 kb,避免启动子干扰
主要输出:
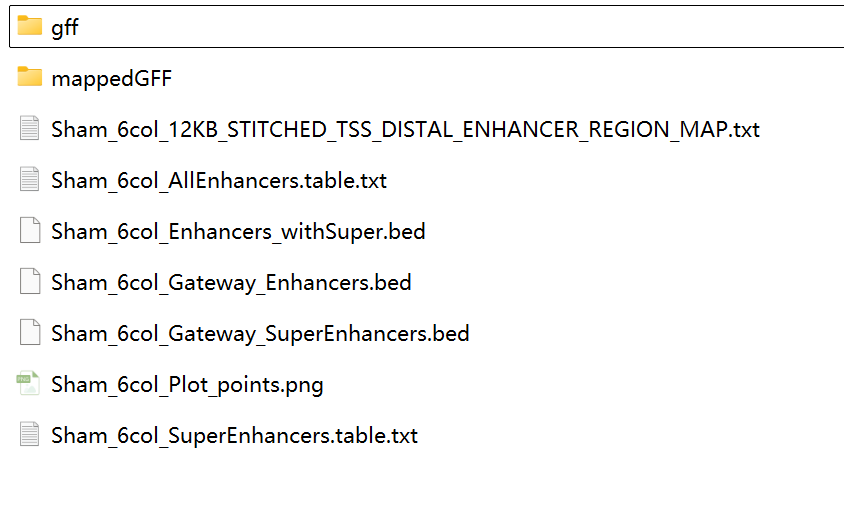
*_SuperEnhancers.table.txt—— 超级增强子列表*_AllEnhancers.table.txt—— 所有增强子(含 TE/SE 标签)*_Enhancers_withSuper.bed—— 可直接导入 UCSC/IGV 浏览器*_Plot_points.png—— rank-plot 拐点图
五、SE 靶基因注释
使用ROSE_geneMapper.py对上一步生成的*_SuperEnhancers.table.txt进行基因注释,代码如下:
python ROSE_geneMapper.py \
-g hg38 \ #参考基因组版本号
-i sample_SuperEnhancers.table.txt \ #输入文件
-o geneMapper #输出文件夹得到:

- TO_GENE.txt —— 每个 SE 邻近基因(50 kb 窗口)
- GENE_TO_ENHANCER.txt —— 基因 ←→ SE 反向索引
后续即可对 SE 关联基因做 GO/KEGG、GSEA、转录因子 motif 富集等分析。
六、非模式物种/自定义基因组
非模式物种/自定义基因组可参考以下步骤:
- 准备
refGene.txt(UCSC genePred 格式)。 - 改名为
mySpecies_refseq.ucsc放入annotation/。 - 修改
ROSE_main.py内GENOME_DICT,添加新基因组键值。 - 其余步骤同上。
七、常见问题
- 染色体命名 —— bam、gff 必须统一带或不带
chr(需要注意参考基因组信息!)。 - Python2 环境 —— 若系统默认 Python3,可通过 conda 创建Python2环境:
conda create -n rose_py2 python=2.7
八、小结
ROSE 以“缝合-排序-拐点”三步法,把 ChIP-seq 峰直接转换成生物学意义明确的超级增强子列表。参考使用上述代码可快速完成从原始 bam 到SE鉴定及注释的全过程。